
Taking Aim at MelanomaUnderstanding oncogenesis at the molecular level offers the prospect of tailoring treatments much more precisely for patients with advanced cases of this deadliest of skin cancers. Pep Karsten / fstop / Corbis They’re lawyers and receptionists, philanthropists and film editors. Some are retired, some just starting families. What they have in common is metastatic melanoma, a cancer that will likely claim their lives in a matter of months. Standard treatments—including removal of tumors and chemotherapy—have failed to halt their advancing disease. But these patients have not given up. All have volunteered to take part in clinical trials designed to test novel therapies that could help rein in their tumor growth and possibly buy them some time. And thanks to two promising new approaches, these patients—and others like them—have reason to be hopeful. The new treatments are more carefully aimed than the massive but indiscriminate hammer blow dealt by standard chemotherapeutics, which have always disappointed in the treatment of melanoma. One approach takes advantage of drugs that have been designed specifically to take down the mutant protein that is inappropriately activated in more than half of all patients with melanoma. Another is a form of immunotherapy that activates T cells that can recognize and attack the tumors. Both have shown an ability to improve the lives of patients with metastatic melanoma, and both are likely to gain FDA approval this year. The results offer hope that someday we may be able to overcome this disease, for which no effective treatments currently exist. Identifying a culprit gene |
Melanoma is the deadliest form of skin cancer. When the disease is caught early, it can be easily treated. But once the cancer spreads, the prognosis is bleak. The standard form of chemotherapy involves treatment with dacarbazine, the only drug approved for treatment of stage IV metastatic melanoma. Dacarbazine interferes with cell growth by chemically modifying DNA, and it is no miracle drug. Only 10 percent of melanoma patients show any response to the drug, and the treatment ultimately has no effect on overall survival.
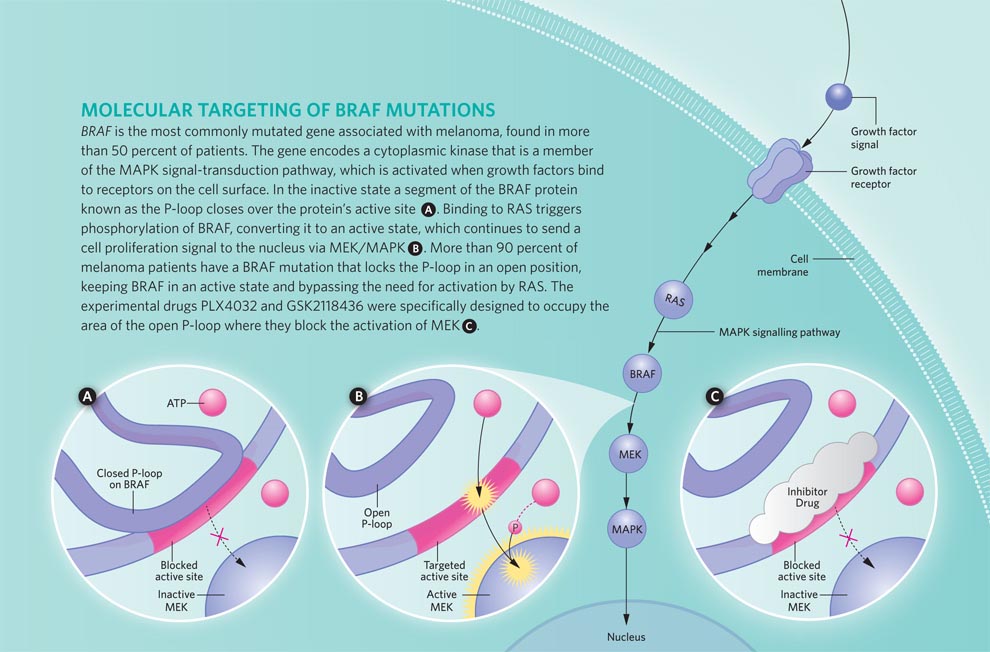
Drugs like dacarbazine are indiscriminate: they batter all cells, whether normal or cancerous. When taking aim at cancer cells, it helps to have a more precisely defined target. In 2002, investigators led by Chris Marshall at the Institute of Cancer Research in London identified such a target. The researchers discovered mutations in a protein kinase called BRAF in more than half of the melanomas they examined.1 These mutations inappropriately activate the kinase, which then turns on a second kinase called MEK. This activational cascade—part of the well-studied MAP kinase signaling pathway—ultimately turns on a suite of genes that mediates cell division. (See “On the MAP,”The Scientist, June 2009.)
The discovery that BRAF activation is a key feature of many melanomas raised the possibility that investigators would be able to identify compounds that would specifically target the aberrant signals that drive this form of cancer. In the years following the identification of BRAF mutations in melanoma, we turned our attentions to sorafenib—a drug that was being tested (and ultimately approved) for the treatment of renal cancer. Sorafenib is a potent inhibitor of the receptors that bind to the angiogenesis-promoting hormone VEGF. It also blocks the activities of a handful of protein kinases, including BRAF.
Unfortunately, we found that sorafenib is a poor inhibitor of BRAF in patients. In 2002 and 2004, I led the clinical trials to assess its ability to act as a single-agent therapeutic for patients with melanoma. In Phase I trials, the maximal dose that could be tolerated by patients was determined, and in Phase II we gave patients as much sorafenib as they could safely handle. But even at the maximal doses, sorafenib showed little to no clinical efficacy in melanoma patients. And biopsies revealed that the drug was not blocking BRAF activity effectively.2,3
Infographic: Molecular Targeting of Braf Mutations
View full size JPG (120 KB) | PDF (560 KB)
Lucy Reading-Ikkanda
View full size JPG (120 KB) | PDF (560 KB)
Lucy Reading-Ikkanda
But we still had high hopes for the targeted approach. Sorafenib was in effect a part-time, accidental BRAF inhibitor—and not a very good one in the end. So the failure of those early trials left the door open for more potent, selective, “professional” BRAF inhibitors to come along. Drugs that were designed specifically to shut down overactive BRAF might be more effective at curbing tumor growth and improving patients’ lives. And with the targeted BRAF inhibitors PLX4032 and GSK2118436, our hopes were realized.4
Attacking BRAF
BRAF is a protein of 766 amino acids. The most common mutation found in melanoma changes the valine (V) at position 600 to a glutamic acid (E). This alteration—dubbed V600E in shorthand—accounts for 90 percent of the mutations in tumor samples analyzed. The next most common mutation, present in a handful of melanomas, changes that same valine to a lysine (V600K). A smattering of other mutation also occur in and around that amino acid hot spot.1
Although targeted BRAF inhibitors have shown promise for malignant melanoma, coming up with more effective combination therapies tops the agenda.
All these mutations function to lock BRAF in its active state. When the protein is inactive, a segment called the P-loop covers the site where ATP binds. When BRAF is activated, the P-loop swings out, exposing this site and allowing the kinase to phosphorylate its target protein, MEK.4 Structural studies, particularly of the V600E form of BRAF, show that mutations repel the P-loop, keeping it from shutting down the kinase activity. (See illustration above.)
The first targeted BRAF inhibitor was designed specifically to recognize and shut down the V600E form of the protein. Developed by scientists at a small biotech company called Plexxikon, the compound—PLX4032—binds to BRAF in its active conformation and prevents it from interacting with its target protein MEK.4 In blocking the activation of MEK, PLX4032 arrests progression into the G1 phase of the cell cycle, halting proliferation. And it also triggers apoptosis—in vitro, in animal models, and in the tumors of patients enrolled in Phase I/II clinical trials. 5
Next to bat
GSK2118436 is a drug developed by the pharmaceutical company GlaxoSmithKline (GSK). Like PLX4032, the GSK compound is a highly selective inhibitor of mutant forms of BRAF. Both bind to the same region of the protein and help hold it in its inactive conformation. But the GSK compound is even more potent than PLX4032: it cuts BRAF’s activity in half at concentrations less than 1 nM. Clinical trials for this potent inhibitor have accepted patients who have any form of the V600 mutant BRAF proteins. Most of those enrolled have the V600E form of mutant BRAF, but a few have the V600K or V600G (valine to glycine) mutation.
Although GSK2118436 has been administered to fewer patients than PLX4032, both drugs have shown similar efficacies in terms of tumor shrinkage. At the maximal dosages, about 90 percent of patients with BRAF mutations show some degree of tumor regression; 60 to 70 percent of patients had tumors that shrank by one-third or more.6 The trials have not gone on long enough to assess either drug’s effect on overall survival, but compared to treatment with dacarbazine, PLX4032 appears to triple the length of time patients have before the disease starts to worsen—from two months with dacarbazine to six months with the new drug.
The GSK compound had the added benefit of actually reducing the size of some of the small brain metastases seen in a subgroup of the patients enrolled in that trial.5 This response was particularly welcome because half of all patients with malignant melanoma will develop brain tumors, which respond particularly poorly to treatment. Once melanoma spreads to the brain, survival can be a matter of weeks, particularly for those with large, symptomatic metastases. So GSK2118436’s ability to shrink brain tumors is being actively pursued.
The trials have been an emotional roller coaster for the patients receiving the drugs—and for the researchers and medical personnel developing and administering them. [See this three-part series, “Target Cancer” featuring Keith Flaherty (The New York Times, Feb 22-24, 2010).] And the benefits seen in the Phase II clinical trials should garner targeted BRAF inhibitors FDA approval sometime in 2011. But the news is not all good. A response rate of 90 percent means that 10 percent of the patients enrolled in the PLX and GSK trials are not helped by these targeted drugs. Their tumors continued to grow during the course of the treatment. In addition, a large percentage of those whose tumors shrank during the first months of treatment will develop drug resistance. Investigators are still exploring what the mechanism of this resistance might be. What they have determined so far is that patients do not seem to accrue any new, reactivating mutations in BRAF itself. Instead, some tumors show mutations that enhance the activity of other kinases in the MAP kinase pathway to which BRAF belongs.7 Other tumors accumulate mutations that activate different signaling pathways which mediate cell proliferation or survival, including that of another oncogenic kinase called phosphoinositide 3-kinase (PI 3-kinase).8
Getting a handle on how tumors can circumvent BRAF inhibitors should give us a better idea of how to attack melanomas that become resistant to these promising new agents. The knowledge could also point the way toward drugs that could be effectively combined with BRAF-targeted inhibitors as a frontline form of therapy for metastatic melanoma to prevent the emergence of these resistance mechanisms. BRAF inhibitors could be given along with targeted inhibitors of other oncogenic kinases, including PI 3-kinase. Another approach would be to combine targeted BRAF inhibitors with an entirely different type of treatment, such as immunotherapy.9
The long haul
Even if targeted molecular therapies like PLX4032 or the GSK compound were 100 percent effective, only half of all patients with melanoma have tumors with BRAF mutations. For the other half, we need to come up with some viable alternatives. Some of the patients have other mutations that might serve the same function as BRAF. For those patients, similar treatment strategies are being developed. But for any patient with melanoma, immunotherapy provides an option that does not appear to depend on specific mutations in the tumor.
Infographic: Boosting T Cell Activation
View full size JPG (205 KB) | PDF (171 KB)
Lucy Reading-Ikkanda
View full size JPG (205 KB) | PDF (171 KB)
Lucy Reading-Ikkanda
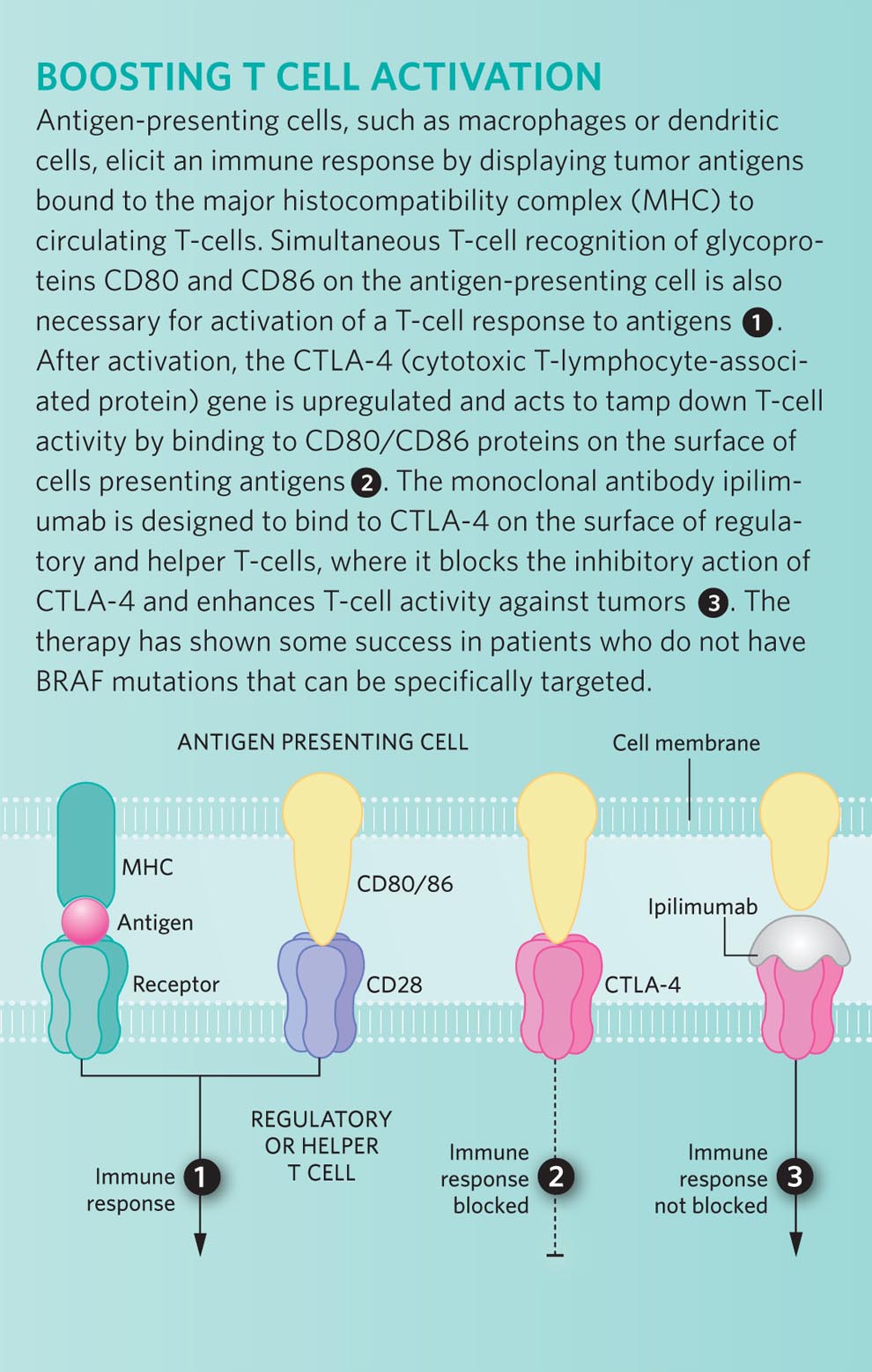
Perhaps the most directed approach would be to prime the immune system to recognize antigens that are specific to melanomas—or at least to the melanocytes that give rise to this cancer. In the mid-1990s, investigators identified a protein that could potentially serve as a target antigen: a membrane-bound glycoprotein called gp100. This protein is present on the surface of cells in the melanocyte lineage, as well as cells of the majority of both primary and metastatic melanomas.
By formulating a vaccine that consists of a small fragment of gp100, researchers hoped they could convince the patient’s immune system to attack tumor cells bearing this melanoma-specific antigen. Unfortunately, the gp100 peptide vaccine has never shown much clinical benefit when given on its own. But animal models and some small clinical trials suggest that when combined with an agent that can also stimulate a patient’s T cells, gp100 could help target immune cells to the tumors that are to be destroyed. One of these T-cell–boosting drugs is ipilimumab.
Ipilimumab (made by Bristol-Myers Squibb) is a monoclonal antibody that recognizes the CTLA4 protein on the surface of regulatory and helper T cells.10 CTLA4 inhibits T-cell activation and helps to promote immunologic tolerance, so blocking this protein should stimulate an immune response and help prime T cells to attack tumors bearing antigenic target proteins.
The results of the first large phase III trial for ipilimumab look promising. In this trial, investigators compared patients who received either a combination of the gp100 vaccine plus ipilimumab to those who received either of the treatments on its own. In this case, the vaccine-only patients served as a “control arm” of the study, since gp100 has not been found to be effective in treating melanoma when administered as a single agent. The results confirmed what had been seen in smaller, nonrandomized studies in which everyone received the drug: about 10 percent of the patients taking ipilimumab—either with or without the peptide vaccine—experienced tumor shrinkage of one-third or more.11 As yet, we do not know why these patients responded to the drug, while others did not. One goal for the future would be to identify what makes these responders unique so that we can target this therapy to the patient population most likely to benefit.
Even though 90 percent of the patients showed no shrinkage, it does not necessarily mean that the drug did them no good. For some of these patients, ipilimumab (again, taken alone or in conjunction with the peptide vaccine) has halted disease progression: their tumors have remained the same size for years—and they are still alive to tell about it. Such a response is unusual for patients with malignant melanoma, a cancer that generally progresses quite rapidly. And among the 10 percent of patients who initially responded to the drug, most continue to benefit. In the randomized trial it was clear that a larger percentage of patients lived for two years after receiving ipilimumab, and from earlier trials some who have been responding for 10 years and counting.
Another large phase III trial—comparing ipilimumab plus dacarbazine to dacarbazine alone—has been going on for several years and we hope the results of that study will corroborate the life-prolonging effects we’ve already seen. But even without that trial, many of us believe that the FDA will look favorably on the results reported thus far, because no other drug tested to date has ever enhanced survival in a phase III trial for malignant melanoma.
More one-two punches
Although targeted BRAF inhibitors have shown promise, none of us is satisfied with what single-agent therapies have accomplished thus far. So for malignant melanoma, coming up with more effective combination therapies tops the agenda. The low-hanging fruit would involve using drugs already developed for targeting other kinases or other pathways in which aberrant activity contributes to the development of melanoma, such as members of the PI 3-kinase pathway.
At the same time, oncologists are migrating toward mixing different types of therapies, such as targeted BRAF inhibitors plus immunotherapy. Our group at Massachusetts General Hospital has produced data supporting the idea that combining those approaches would likely benefit our patients. We have found that melanoma cells exposed to BRAF inhibitors become more densely decorated with surface antigens such as gp100. In the absence of treatment, these cancer cells somehow suppress expression of their melanocyte-specific antigens, which presumably helps them to escape immune surveillance. And BRAF inhibitors seem to reverse this suppression. Working with melanoma biopsies taken before and after therapy with BRAF inhibitors, we have found that treated tumor cells display more melanocyte- specific antigens and are more easily recognized by T cells harvested from the patient.9
Of course, convincing pharmaceutical companies to make their drugs available for such combination trials can in itself be challenging. First, the specificity of each treatment could mean that any combination would further fractionate the population of patients for whom the treatment would work. For example, more than half of patients with melanoma have a mutation in BRAF. And a smaller percentage of those will have a BRAF mutation combined with a companion mutation in another kinase for which an inhibitor is available or can be developed. Drug companies can be loath to move forward with therapies for which the market promises to be small.
Pharmaceutical companies also shy away from coadministering drugs because of the fear that a drug combination could produce a unique toxicity—something that would not be an issue with either drug alone. Such a reaction could kill a compound that’s still in development—even if that drug would be perfectly safe and perhaps effective when taken as a single agent.
Cancer is a complex disease, and developing effective treatments is more complex still. For the benefit of patients, my colleagues and I contend that the whole drug development industry needs to recognize this complexity and incentives need to be provided to companies that are willing to take on the challenge of formulating new therapies—and combinations of therapies—to tackle this disease. The science is out there to support these approaches. Now, we as physicians need to show as much determination as our patients in our ongoing search for the drugs—or combinations of drugs—that will allow people with malignant melanoma to return home and go on living their lives.

Andrzej Tokarski / ISTOCKPHOTO.COM
A DIET OF PILLS
There comes a point in any trial when investigators agree to pause and take stock of the situation. In Phase I trials, for example, we typically stop to determine whether we’re seeing any side effects that would warrant discontinuing the trial—or whether we’re seeing any effects that would suggest we move forward and expand the number of patients receiving the drug. In the case of PLX4032, developed at a small California biotech company called Plexxikon, we ran into a different problem: we hit a plateau in terms of bioavailability. In other words, we could increase the dosage of drug the patients were receiving, but we couldn’t get their blood concentrations high enough.
On the one hand, we could have opted to push forward, persuading our patients to swallow even more of the poorly absorbed pills. But there are drawbacks to that approach. First, overloading people’s digestive systems with drugs can produce local irritation, resulting in diarrhea and the like. Although these conditions are not life-threatening, they can make it difficult for patients to tolerate the therapy—not a good outcome for any new drug. And even if the megadoses don’t cause intestinal distress, a high pill count can cause problems down the road. Patients in Phase I trials are a highly motivated cohort, willing to serve as guinea pigs on drugs that have never before been tested in humans. But what if patients in later trials are not as willing to do whatever it takes? If your drug only works at the highest dose, but your patients are not willing to swallow more than 30 pills a day to achieve that dose, then the trial will fail to show efficacy, and a potentially useful drug could be permanently shelved.
Instead, we voted to pause—to wait as long as it would take to derive a better formulation. If that formulation could not be found, we would consider walking away, trying to find another agent that could achieve the concentrations that would be needed to really take down the mutant BRAF. Fortunately, Plexxikon and Roche (the company’s new partner in developing PLX4032) were willing and able to rework the drug into a form that boosted blood concentrations higher. Even then, we saw no significant side effects, so we increased the dosage—to the point that people were taking 14 capsules, twice a day. That was a steady diet of pills.
Since then, the drug has been made even more concentrated, so fewer, smaller capsules will yield the same effect. It’s important to remember that we are working with people here—people we wish to help. So we have to respect their limits. In doing so, we not only benefit the patients in our trial, but all patients who will ultimately benefit from receiving an effective treatment in the future.
Editor's Note (4th April): Just prior to the publication of this article, the FDA approved ipilimumab for use in advanced melanoma patients. It was shown to lengthen the life spans of 20-30% of advanced melanoma patients participating in a clinical trial of the drug. Some patients lived several years after their initial diagnosis.
F1000 Member Keith T. Flaherty is Director of Developmental Therapeutics at Massachusetts General Hospital Cancer Center in Boston.

AlphaMed Press, publisher of THE ONCOLOGIST® and THE ONCOLOGIST COMMUNITYSM website, grants The Scientist permission to provide this video link to Keith T. Flaherty’s lecture, "BRAF: the first prevalent oncogene target to show single-agent responsiveness," at THE ONCOLOGIST COMMUNITY.
1. H. Davies et al.,“Mutations of the BRAF gene in human cancers,” Nature, 417:949-54, 2002. Free F1000 Evaluation
2. K.T. Flaherty et al.,“Phase I/II, pharmacokinetic and pharmacodynamic trial of BAY 43-9006 alone in patients with metastatic melanoma,” J Clin Oncol, 23(16s): abstract 3037, 2005.
3. T. Eisen et al., “Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis,” Br J Cancer, 95:581-86, 2006.
4. G. Bollag et al., “Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma,”Nature, 467: 596-99, 2010. Free F1000 Evaluation
5. R. A. Kefford et al., “Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors,” J Clin Oncol, 28(15s): abstract 8503, 2010.
6. K. T. Flaherty et al., “Inhibition of mutated, activated BRAF in metastatic melanoma,” N Engl J Med, 363:809-19, 2010. Free F1000 Evaluation
7. C.M. Johannessen et al., “COT drives resistance to RAF inhibition through MAP kinase pathway reactivation,”Nature, 468:968-72, 2010. Free F1000 Evaluation
8. R. Nazarian et al., “Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation,”Nature, 468:973-77, 2010.
9. A. Boni et al., “Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function,” Cancer Res, 70:5213-19, 2010. Free F1000 Evaluation
10. J.S. Weber et al., “Phase I/II study of ipilimumab for patients with metastatic melanoma,” J Clin Oncol,26:5950-56, 2008.
11. F.S. Hodi et al., Improved survival with ipilimumab in patients with metastatic melanoma, N Engl J Med,363:711-23, 2010. Free F1000 Evaluation








